Get started
Install & setup
BulkSeq Studio is free and runs on Windows and Linux. You download one file, launch it, and let the app set up the bioinformatics tools for you the first time. No coding, and on Windows no administrator account for the install itself.
Windows
Two downloads on the Releases page, pick either:
- Installer —
BulkSeqStudio-Setup-0.16.0.exe. A per-user install that needs no administrator rights; afterwards you launch it from the Start Menu. - Portable —
BulkSeqStudio-Portable-0.16.0.zip. Unzip it anywhere and double-clickBulkSeqStudio.exe. Nothing is installed.
To update, run a newer installer: it detects the existing install and offers to update or uninstall first.
Linux
Download from the same Releases page and pick either:
- AppImage — a single self-contained file. Make it executable, then run it; nothing to extract or install. To update, download the newer file or run AppImageUpdate on it (the build ships zsync update info).
- Tarball — extract it and run the bundled launcher
BulkSeqStudio.
Both bundle the interface, so you do not need system Python. They are built on Ubuntu 24.04 and need glibc 2.39 or newer.
What is WSL2, and why does Windows need it?
The bioinformatics tools that do the analysis (alignment, counting, DESeq2, enrichment) are Linux programs. On Windows, BulkSeq Studio runs them inside WSL2 — the Windows Subsystem for Linux, a lightweight Linux that Windows runs quietly in the background. You never open a Linux terminal yourself. The window you see is only the graphical interface; the actual computation happens in a managed micromamba environment inside WSL2. The app can enable WSL2 and install that environment for you from its setup screen, so you do not configure anything by hand.
On Linux there is no WSL: the same interface runs the same pipeline directly in a local environment, and the results are identical.
Install and first run, step by step
-
Download and open the app
Grab the Windows installer or portable ZIP (or, on Linux, the AppImage or tarball) from the Releases page, then launch BulkSeq Studio. The per-user Windows install does not ask for an administrator password.
-
Let the Environment setup screen check your machine
On first launch (and any time later from Check Environment on the Project tab), a readiness screen checks the pieces it needs and shows each one as Ready or Action needed, with an install button when something is missing.
-
Install anything marked "Action needed"
Work top to bottom: click each Install… button, then press Re-check to refresh the statuses. The downloads run inside your own WSL user account with no sudo password. Show details / log expands the full setup log if a step needs attention.
-
Click Continue when the header reads "4 of 4 ready"
Once every row is Ready, the screen is done. You can already create projects, edit metadata, and build configs while the tools finish installing; only starting a pipeline run needs them.
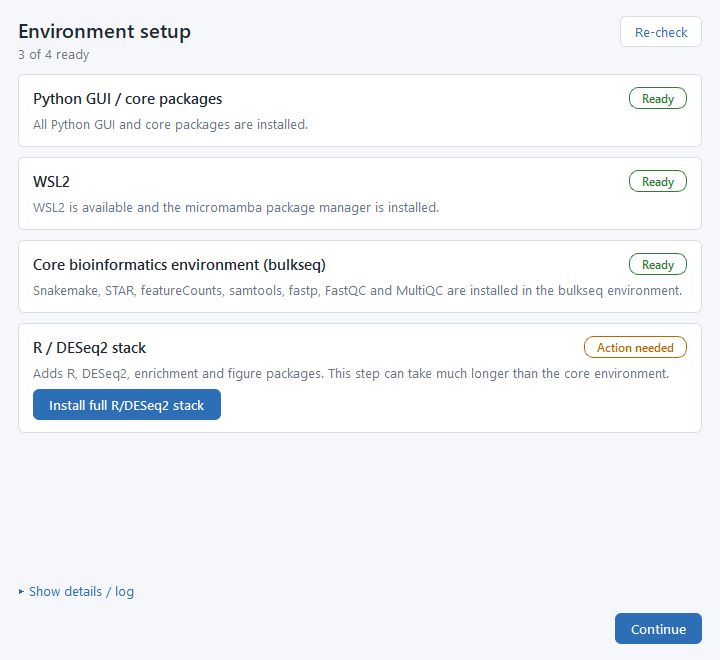
The four readiness rows (Windows first run)
- Python GUI / core packages — the libraries the interface itself needs.
- WSL2 — the lightweight Linux the pipeline runs inside, plus the micromamba package manager. If WSL2 is not yet enabled, this single step opens an Administrator PowerShell window once (Windows requires elevation only to turn the feature on), and you may be asked to reboot.
- Core bioinformatics environment ("bulkseq") — Snakemake, the three aligners (STAR, HISAT2, Salmon), featureCounts, samtools, fastp, FastQC, and MultiQC. The download takes a few minutes.
- R / DESeq2 stack — R with DESeq2, the enrichment packages, and the figure toolchain. This is the largest, slowest install and can take a while; you only do it once.
On Linux the WSL2 row does not apply — the pipeline runs locally and those controls are hidden.
Raising the WSL2 memory limit (Windows)
On Windows the pipeline runs inside WSL2, which by default may use only about half of your host RAM. That VM cap — not the Windows total — is what memory-heavy steps run against (STAR indexing and alignment on large genomes, or DESeq2 on big count matrices). The easiest way to raise it is the Edit WSL2 memory / CPU limits… button on the Resources tab, which writes the caps and can restart WSL to apply them. To do it by hand instead, create or edit %UserProfile%\.wslconfig (a plain text file in your user folder), add a [wsl2] section, then apply it with wsl --shutdown in PowerShell before reopening the app:
[wsl2]
memory=48GB # cap the VM at 48 GB (default is ~50% of host RAM)
processors=16 # optional: limit WSL2 to 16 logical processorsSee Microsoft's Advanced settings configuration in WSL ↗ for every .wslconfig option. On Linux there is no cap to configure — the pipeline runs natively with your machine's full RAM.
Your first analysis
Quick start
From a public study to differential expression, enrichment, and an interactive protein network, without writing any code. The example below follows the most common path: paste study accessions, let the app fetch the data, and run.
-
Create or open a project
On the Project tab, make a new project folder. The app scaffolds the
config/folder, a copy of the Snakemakeworkflow/, and a provenance manifest. On Windows, click Use WSL filesystem when prompted so the project lives on the fast Linux disk rather than aC:\drive. -
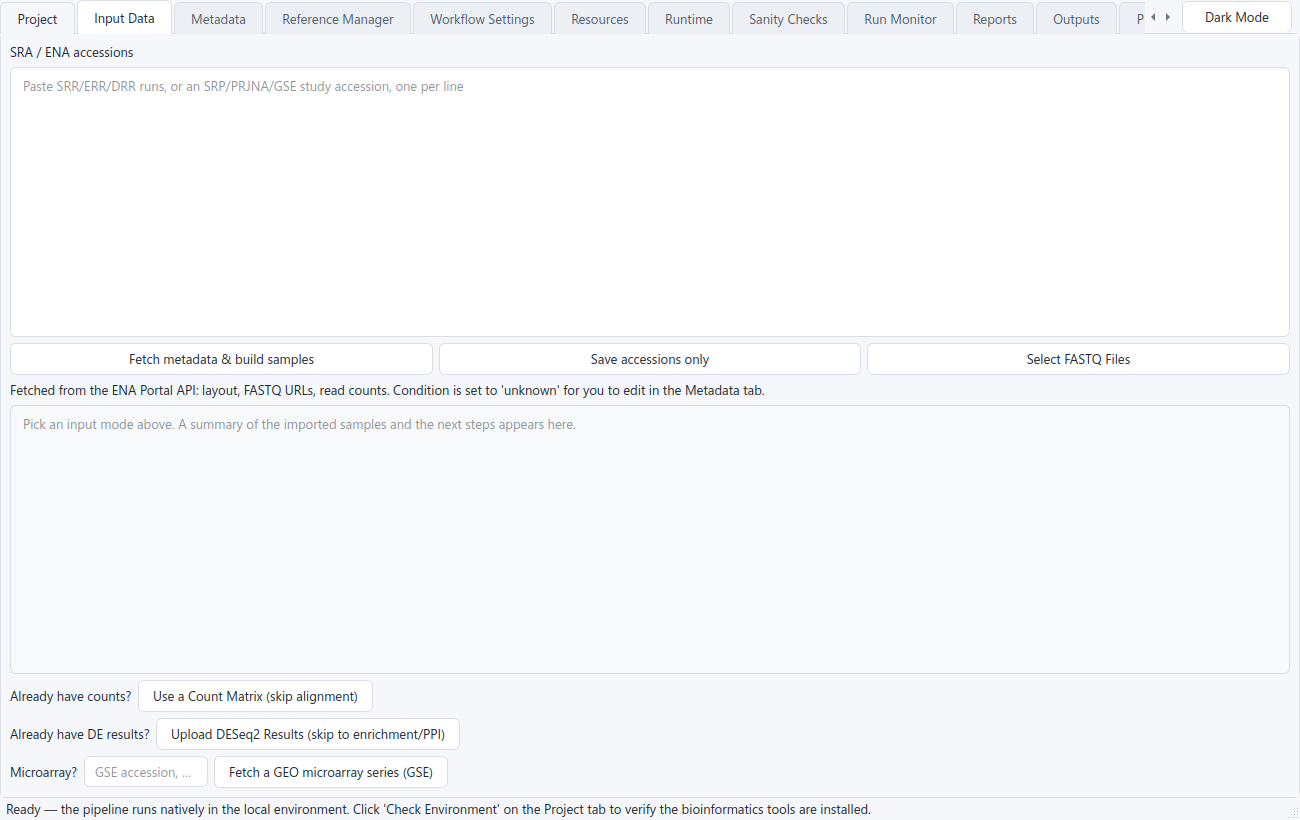
Choose an input
On the Input Data tab, pick how your data comes in. The simplest is to paste SRR / SRP / PRJ accessions — or an RNA-seq GSE series, which is resolved to its SRA runs — and click Fetch metadata & build samples; the ENA fetch reads the layout, FASTQ URLs, and read counts and builds the sample sheet for you. You can also select local FASTQ files, or skip alignment entirely with Use a Count Matrix, Upload DESeq2 Results, or a GEO microarray series (GSE).
-
Confirm the metadata and contrast
On the Metadata tab, the samples appear in an editable table. The
conditioncolumn is pre-filled with a group suggested from the fetched metadata (a GEO characteristic such as genotype or treatment, or a sample-title group) — confirm it or edit it, and check that the layout (paired or single-end) is correct. These groups define the comparison DESeq2 will run. -
Pick a reference and analysis options
On the Reference Manager tab, choose your organism from the Ensembl / NCBI RefSeq catalog (or import a custom genome and annotation), then click Use Selected Preset. A reference is required before a run will start. On the Workflow Settings tab, set the trimmer, aligner, and rRNA tool (leave the aligner on STAR if unsure), pick the differential-expression engine, build the DESeq2 design and contrast (a Design helper can compose the formula from your metadata), the
alphaand|log2FC|thresholds, and how to handle organellar genes. The defaults are sane, so most first runs only need the contrast confirmed. -
Start the run and watch the Run Monitor
Clear any flagged items on the Sanity Checks tab, then go to Run Monitor and click Start Run. The progress bar and a plain-language current phase (Downloading, Aligning, DESeq2, and so on) track the pipeline above the raw log. Use Stop to cancel, Resume to continue an interrupted run, and Unlock if a run was interrupted hard.
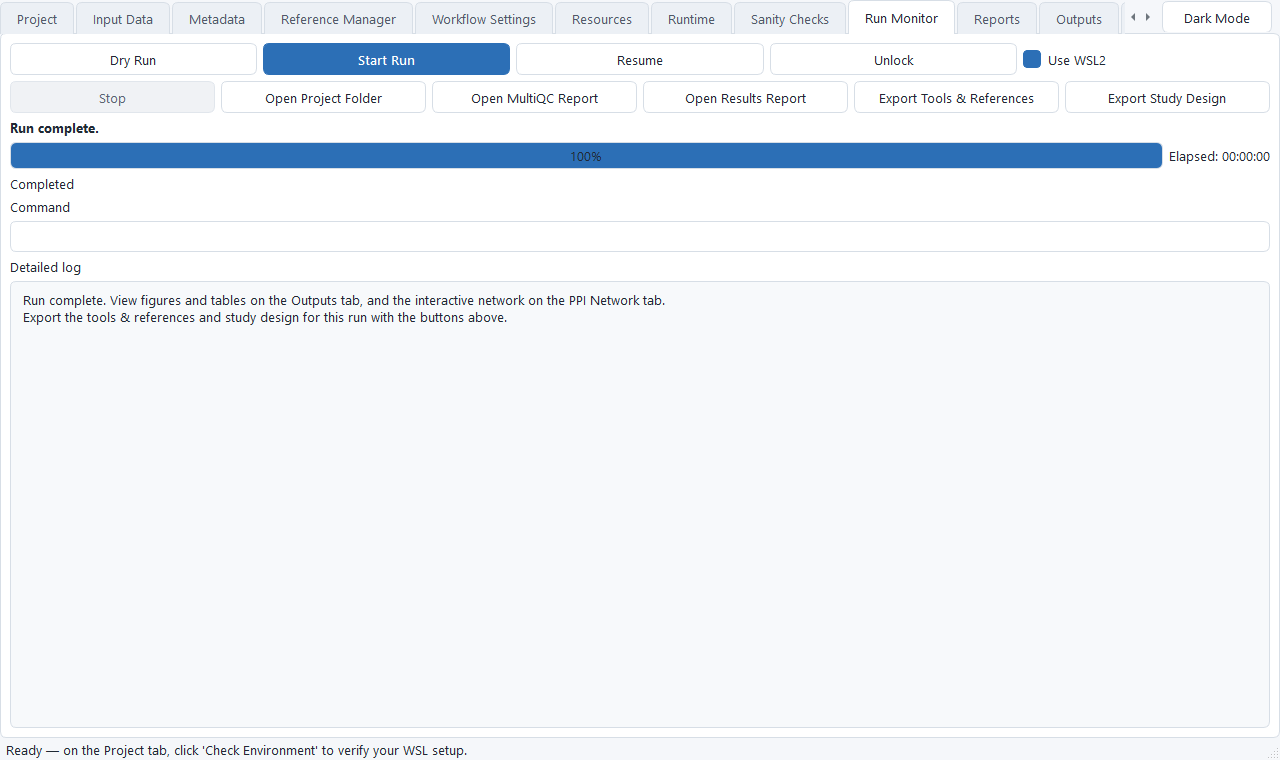
The Run Monitor after a finished run. The Export Tools & References and Export Study Design buttons save the exact tool versions, reference, and study design for that run. -
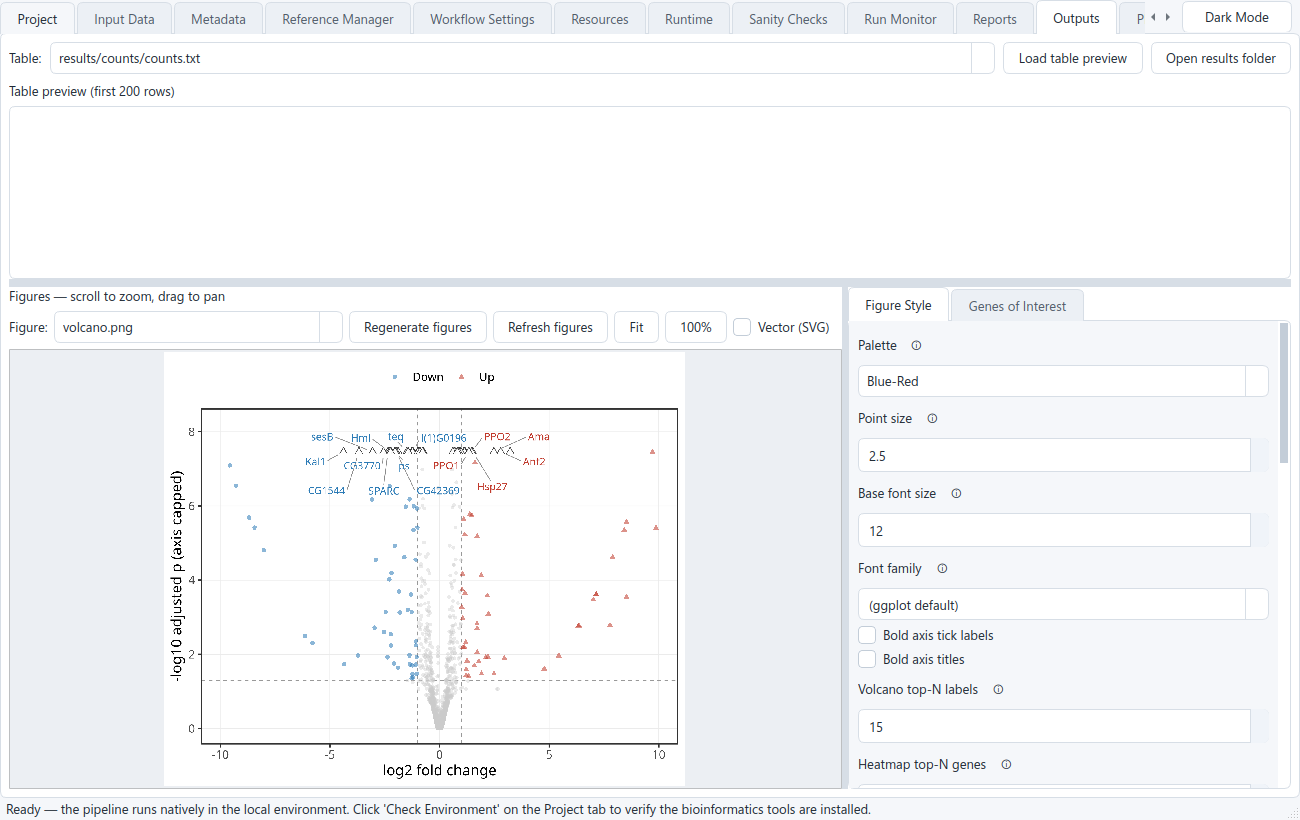
Read the results
When the run finishes, the Outputs tab lets you browse the count matrix and DESeq2 table and view the figures (PCA, volcano, MA, heatmaps); toggle Vector (SVG) for a crisp preview at any zoom. Open the PPI Network tab to explore the STRING protein network interactively, and click Open MultiQC Report on the Run Monitor for the read-quality summary. A self-contained
results_report.htmlgathers the figures, top genes, enrichment, and provenance into one shareable file. Export the tools & references and study design files to capture the run's provenance.
Five ways in
Input modes
BulkSeq Studio meets your data wherever it already is. You can start from raw reads, from public accessions, from a count table, from a finished differential-expression table, or from a GEO microarray series. The mode you choose decides which steps run and which outputs you get; everything downstream of the entry point is shared, so the figures and enrichment look the same no matter how you got there.
1. SRA / ENA accessions
Paste SRR / SRP / PRJ accessions and the app fetches the ENA metadata, builds the sample sheet (layout, FASTQ URLs, read counts), and downloads the reads for you.
Unlocks the full pipeline: FastQC / MultiQC, trimming, alignment, gene counting, DESeq2, enrichment, figures, and the PPI network.
2. Local FASTQ files
Already downloaded your reads? Point the app at FASTQ files on disk. Single-end or paired-end, detected automatically from the sample sheet; both layouts run the full alignment route.
Same full pipeline as the accession route, minus the download step: QC, trimming, alignment, counting, DESeq2, enrichment, figures, PPI.
3. Count matrix (skip alignment)
Upload a gene-by-sample counts table (featureCounts output or any TSV/CSV) to skip download, QC, and alignment.
Goes straight to DESeq2, then figures, enrichment, statistics, and the PPI network. Use this when alignment is already done elsewhere.
4. DESeq2 results table (bring your own)
Upload a finished differential-expression table (the format DESeq2::results() writes). Required columns are gene_id, log2FoldChange, and padj; the up- and down-regulated lists are derived from your significance thresholds.
No FASTQ, alignment, counts, or DESeq2 step. Produces functional enrichment, the volcano / MA / p-value figures, the STRING PPI network, the MSigDB set-overlap test, and a preranked .rnk. Outputs that need per-sample counts (PCA, sample-correlation, count heatmaps, the Wilcoxon test, the normalized-expression export) are skipped.
5. GEO microarray series (GSE)
Enter a GEO series accession. The app ingests the normalized intensities (GEOquery series matrix, or RMA from raw Affymetrix CEL), maps probes to gene symbols, and runs limma differential expression into a DESeq2-shaped results table.
From there it runs the same figures, enrichment, genes-of-interest, and networks as the RNA-seq routes. RNA-seq series pasted here are redirected to the SRA box.