What you get
Outputs
A finished run leaves a project folder of result tables, publication figures, a quality report, enrichment and network files, and a record of exactly how it was produced. You browse all of it inside the Outputs tab; every file is a plain CSV, PNG, SVG, or standard network format you can open or share.
Result tables
The differential-expression results come out as tab-friendly text files you can open in Excel or R:
- DESeq2 / limma results table — every gene with its log2 fold-change, p-value, adjusted p-value, gene symbol, and biotype.
- Up- and down-regulated gene lists — the significant genes split by direction, using your
alphaand|log2FC|thresholds. - Normalized expression matrix — DESeq2 normalized counts (or log2 intensities for microarray) as a gene-by-sample CSV.
- Unchanged / equivalence list — genes flagged as genuinely unchanged by the TOST equivalence test, separate from "not significant".
- Preranked
.rnk— a stat-ranked gene list for running GSEA elsewhere.
Figures
Each figure is written in two forms: a raster PNG for documents and slides, and a vector SVG that stays crisp at any zoom (toggle Vector (SVG) in the viewer to preview it). The Figure Style editor restyles and regenerates them without re-running alignment or DESeq2.
- Volcano and MA plots.
- PCA and sample-distance plots.
- Top-DEG and genes-of-interest heatmaps.
- Enrichment dotplots (GO and KEGG), GSEA running-score and ridgeplots, gene-concept and term-similarity networks.
- An optional GSVA pathway-activity heatmap from your own gene sets.
- A raw p-value histogram and, for RNA-seq, dispersion / Cook's-distance / library-size diagnostics.
Quality report
The MultiQC report gathers FastQC, trimming, alignment, and (when enabled) FastQ Screen contamination and RSeQC read-distribution / gene-body-coverage statistics for every sample into one HTML page. Open it from the Open MultiQC Report button on the Run Monitor.
Self-contained HTML report
Every run also writes a single-file results_report.html that inlines the key figures, the top genes, the enrichment results, and the run's provenance into one document with no external assets. Because everything is embedded, you can email it, drop it in a shared folder, or archive it and it still renders on its own — a quick way to hand the result to a collaborator without shipping the whole project folder.
Enrichment tables and networks
- Enrichment tables — the GO, KEGG, and custom gene-set over-representation and GSEA results as CSV, run separately on the up- and down-regulated sets.
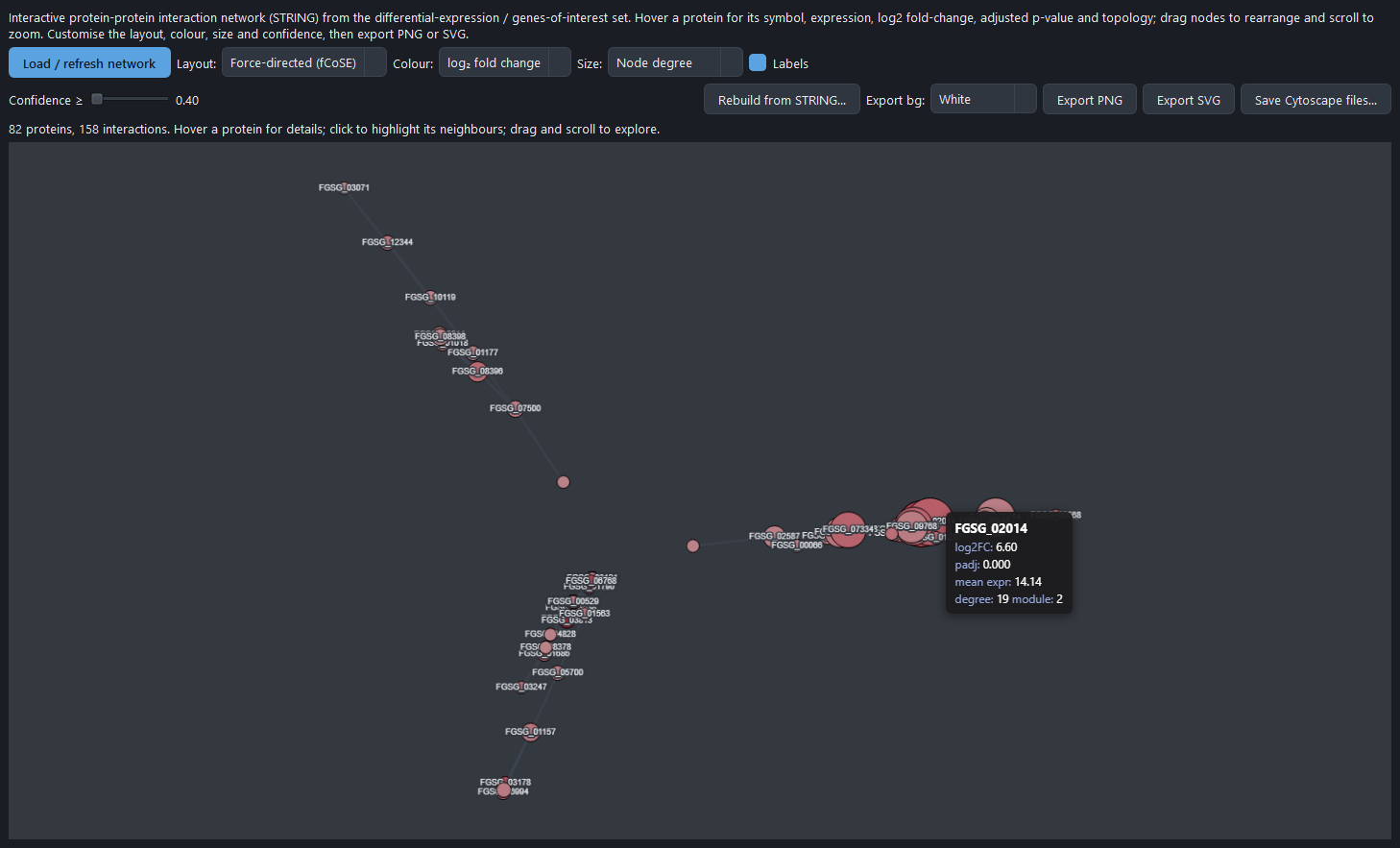
- STRING network files — the protein-interaction network exported as GraphML, SIF, and cytoscape.js JSON (cyjs) for editing in Cytoscape.
- Interactive PPI Network tab — the same network rendered live in the app (see below).
Genes of interest
Supply a gene list and the app generates a focused z-scored heatmap, a per-condition expression panel, and a counts table from the existing run, plus a STRING network for those genes when PPI seeding is set to the list. No re-analysis is needed.
Run provenance
When a run finishes, the Run Monitor lets you save two records of how it was produced:
- Tools & references — the tool and R/Bioconductor package versions, the reference genome and annotation with source URLs and MD5, and the enrichment database codes.
- Study design — the samples, conditions, layout, design formula, and contrasts.
sessionInfo, so a run can be reproduced later.